An initial velocity is assigned to each atom, and Newton's laws are applied at the atomic level to propagate the system's motion through time (see `Classical and Quantum Mechanics - in a Nutshell' above). Thus, dynamical properties such as time correlation functions and transport coefficients (e.g., diffusion constants, bulk viscosities) can be calculated from a sufficiently long MD trajectory.
Once again, Newton's second law is:
![]() , where
, where
![]() is the sum of all forces acting on atom i that results in its acceleration
is the sum of all forces acting on atom i that results in its acceleration ![]() .
The acceleration is the second derivative of the position with respect to time:
.
The acceleration is the second derivative of the position with respect to time:
![]() . In words, it is the rate of change of the velocity
. In words, it is the rate of change of the velocity ![]() , which in turn,
is the rate of change of the position
, which in turn,
is the rate of change of the position ![]() .
.
The `Leap Frog' algorithm is one method commonly used to numerically integrate Newton's second law.
We obtain all atomic positions ![]() at all times
at all times ![]() and all atomic velocities
and all atomic velocities ![]() at intermediate
times
at intermediate
times ![]() . This method gets its name from the way in which positions and velocities are calculated in an alternating
sequence, `leaping' past each other in time:
. This method gets its name from the way in which positions and velocities are calculated in an alternating
sequence, `leaping' past each other in time:
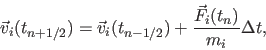
The energy of an isolated system (as opposed to, for example, one in contact with a thermal bath) is conserved in nature,
but it may not be in simulations. Energy conservation can be violated in simulations because of an insufficiently short
integration time step ![]() , an inadequate cutoff method applied to long-range (electrostatic and Lennard-Jones) forces, or
even bugs in the program. Of course, energy conservation alone is not sufficient to ensure a realistic simulation.
The realism of the dynamics trajectory depends on the empirical potential energy function
, an inadequate cutoff method applied to long-range (electrostatic and Lennard-Jones) forces, or
even bugs in the program. Of course, energy conservation alone is not sufficient to ensure a realistic simulation.
The realism of the dynamics trajectory depends on the empirical potential energy function ![]() , the treatment of long-range
forces, the value of
, the treatment of long-range
forces, the value of ![]() , etc.
, etc.