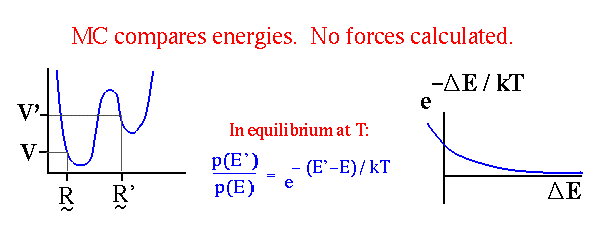 |
|---|
| Monte Carlo makes use of Boltzmann probabilities, not forces. |
When the potential energy V and observables to be calculated from the simulation are velocity-independent (as is typical),
an MC simulation need only compare potential energies V, not total energies E
(see `Calculating Equilibrium Averages' above).
Two conformations, ![]() and
and
![]() , are compared and updated as shown below [13].
, are compared and updated as shown below [13].
![]() is a random number uniformly distributed on [0,1].
is a random number uniformly distributed on [0,1].
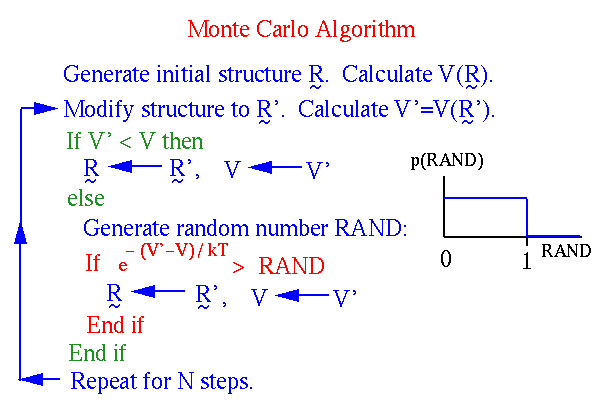 |
|---|
| Metropolis Monte Carlo. |
For simple systems, the structural modifications are often tuned so that about 50% of the
![]() conformations are accepted. For macromolecular systems,
this acceptance ratio can be much smaller, e.g. when dihedral angles are modified by large amounts.
It is then generally expedient to bias the random moves in favor of known structural preferences such as side chain rotamers
(`biased probability Monte Carlo'). In searching for low-energy local minima, it can be advantageous to
minimize the energy before evaluating the energy
conformations are accepted. For macromolecular systems,
this acceptance ratio can be much smaller, e.g. when dihedral angles are modified by large amounts.
It is then generally expedient to bias the random moves in favor of known structural preferences such as side chain rotamers
(`biased probability Monte Carlo'). In searching for low-energy local minima, it can be advantageous to
minimize the energy before evaluating the energy ![]() (`Monte Carlo-minimization', or MCM [14]).
Simulated annealing has also been performed prior to accepting or rejecting the new conformation in a `Monte Carlo-minimization/annealing' (MCMA)
protocol [15]. Because explicit water molecules can hinder the acceptance of new conformations, Monte Carlo (or MCM, MCMA) simulations
of macromolecules generally use an implicit model of solvation, as in references [15,16]. That is, a term is added to the empirical potential energy function
that mimics the effects of water and, in some cases, counter ions.
(`Monte Carlo-minimization', or MCM [14]).
Simulated annealing has also been performed prior to accepting or rejecting the new conformation in a `Monte Carlo-minimization/annealing' (MCMA)
protocol [15]. Because explicit water molecules can hinder the acceptance of new conformations, Monte Carlo (or MCM, MCMA) simulations
of macromolecules generally use an implicit model of solvation, as in references [15,16]. That is, a term is added to the empirical potential energy function
that mimics the effects of water and, in some cases, counter ions.